|
|
DUODENUM - IHC
|
Pending normal tissue analysis |
|
|
|
|
|
DUODENUM - RNAseq
? »
|
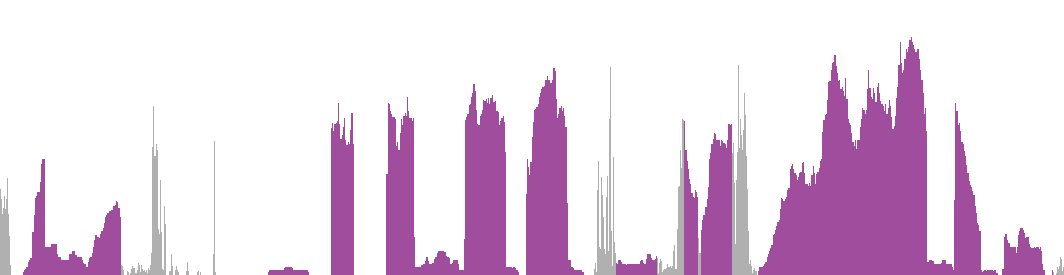
The RNAseq details section shows detailed information about the individual samples used for the transcript profiling and results of the RNAseq analysis.
On top the transcript layout for all coding and non-coding transcripts of the gene, with introns scaled down by a factor 20, is shown. Untranslated regions (UTRs) of protein-coding transcripts are colored gray.
Information about each individual sample is listed below, including gender, age, a tissue section image and estimated fractions of cell types. FPKM (Fragments Per Kilobase of exon per Million fragments mapped) values give a quantification of the gene abundance which is comparable between different genes and samples. The FPKM is calculated as the sum of FPKMs of all protein-coding transcripts.
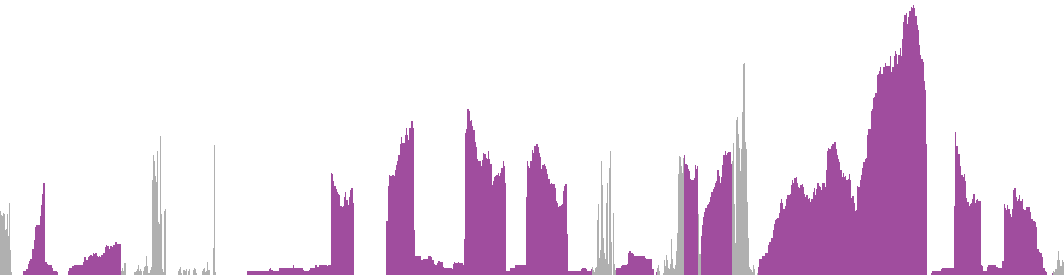
The plots show the mapped read coverage along all exons and introns of the gene, thereby revealing which exons are expressed and at what relative levels. The levels are normalized by total library size and scaled to fit the highest peak of all samples to the height of the plot. Thus these plots should not be used to compare abundances between different samples or genes, but rather as an indication of which transcripts are expressed and as a quality control.
|
|
|

Female, age 77
|
Sample 1
FPKM: 24.7
Cell types% Glandular cells: 45Smooth muscle cells: 10Other cell types: 45
|
|
Male, age 68
|
Sample 2
FPKM: 17.3
Cell types% Glandular cells: 50Smooth muscle cells: 5Other cell types: 45
|
|
|
|
|
|
|